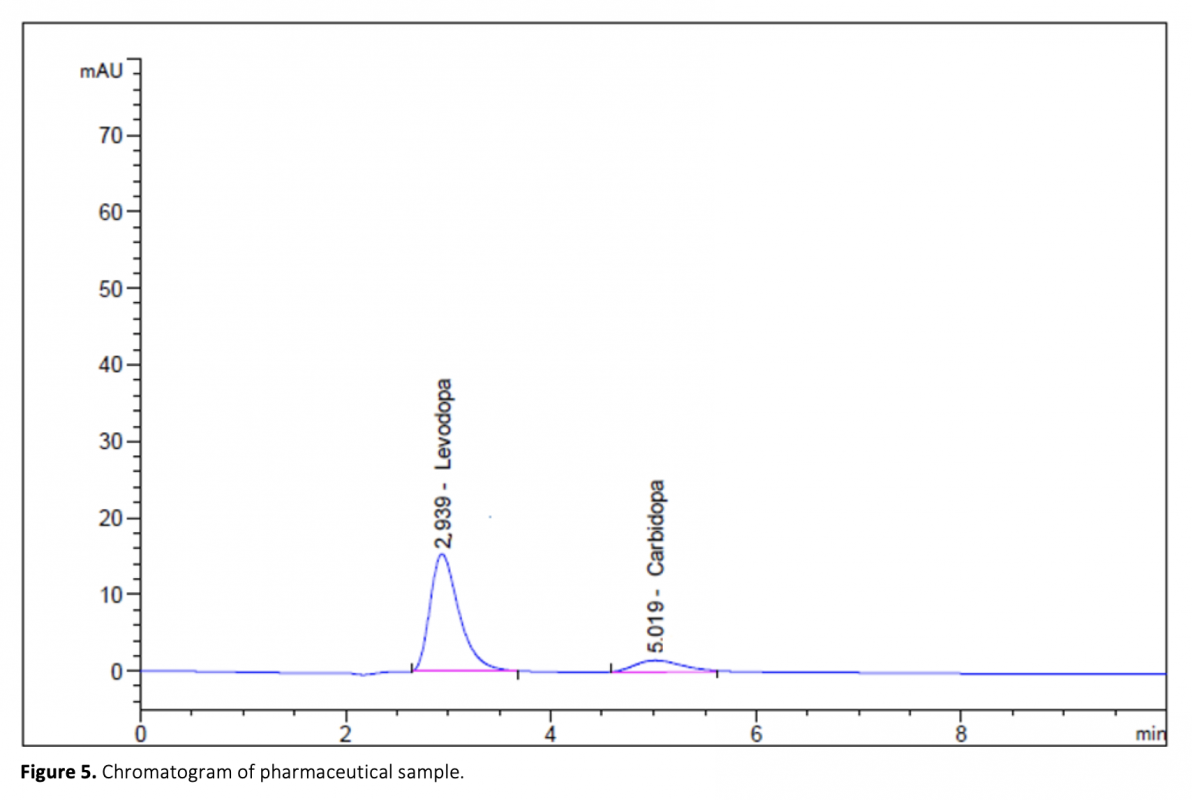
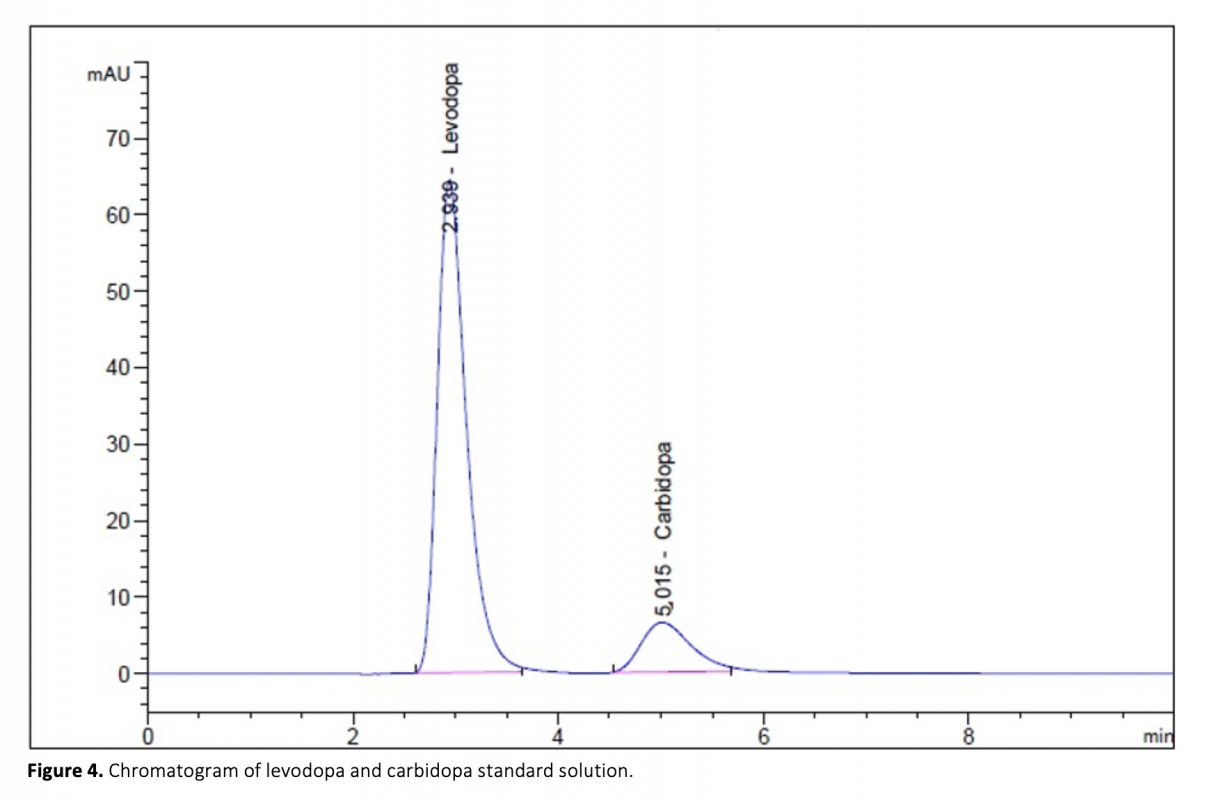
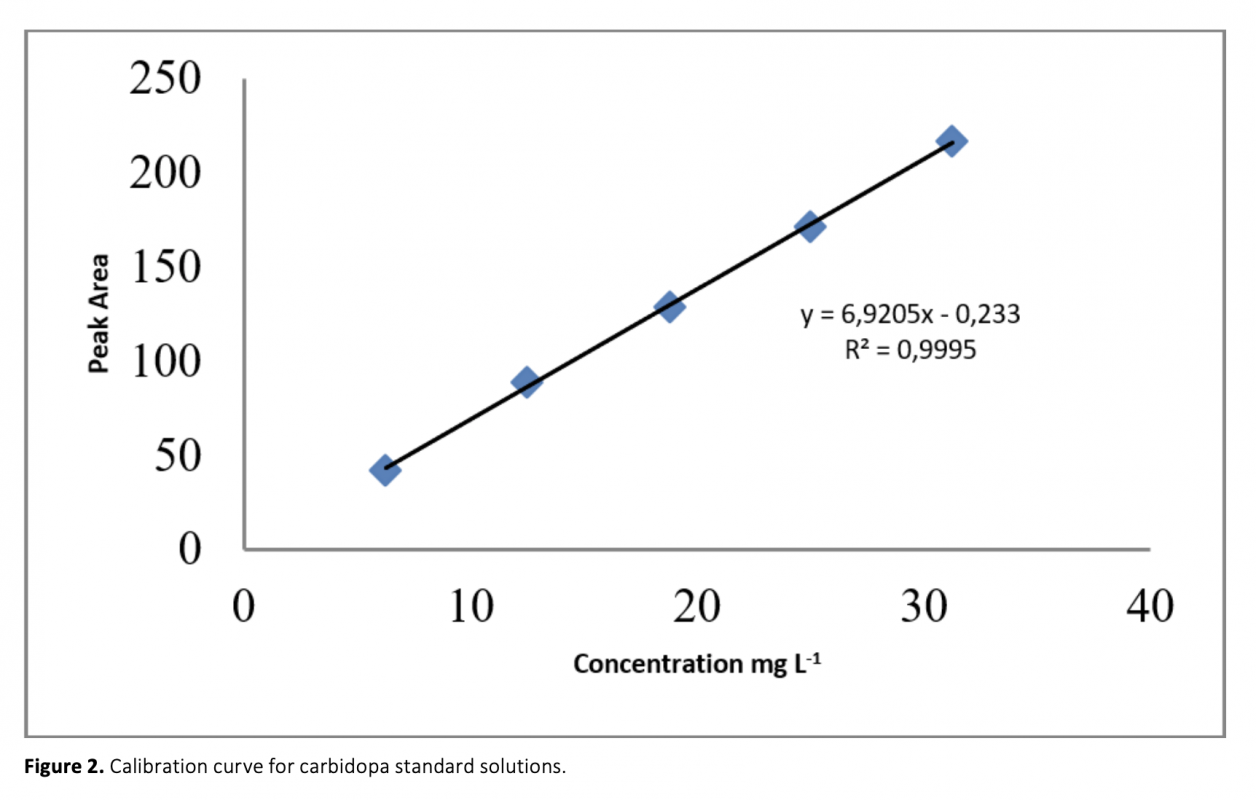
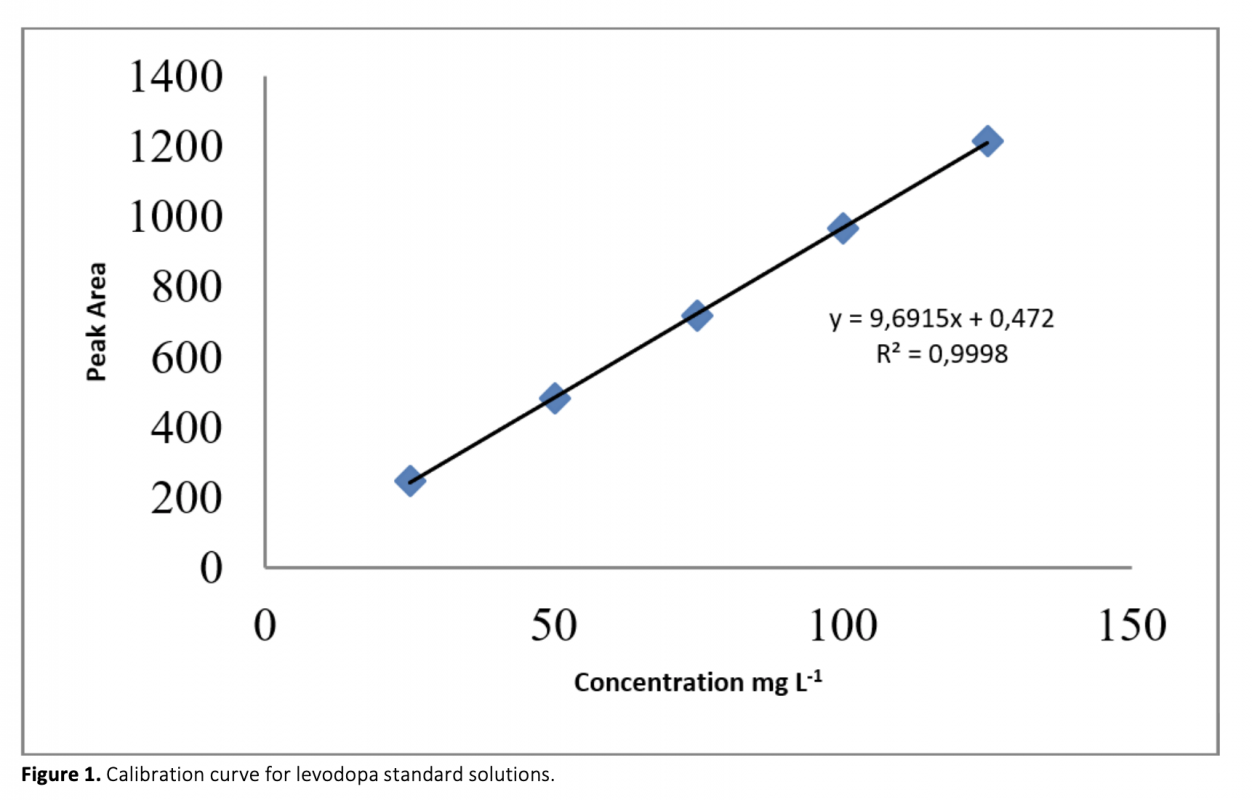
Aconvenient and simple high performance liquid chromatography method for simultaneous separation, determination and identification of levodopa and carbidopa in pharmaceutical formulation has been developed. The analysis was carried out using Ace C18 (4.6 x 250 mm, 5 μm) column, and the separation was performed using a mobile phase consisting of 50 mM KH2PO4 (pH 2.3) pumped at a flow rate of 1.2 mL min-1 with UV detection at 280 nm. The method has been successfully validated on the basis of the International Conference on Harmonization (ICH) acceptance criteria. The method is selective, since no interferences corresponding to these analytes were found at retention times. Retention times for both drugs were 2.939 min and 5.018 for levodopa and carbidopa, respectively. The method was validated and shown to be linear in the concentration range of 25-125 μg mL-1 and 6.25-31.25 μg mL-1 for levodopa and carbidopa, respectively. The method exhibited good linearity (R2 > 0.999) over the assayed concentration range and showed good intra-day and inter-day precision. The limit of detection (LOD) and limit of quantification (LOQ) were determined based on standard deviation of the intercept and the slope of the calibration curve. LOD and LOQ values were 0.70 μg mL-1 and 2.13 μg mL-1 for levodopa and 1.31 μg mL-1 and 3.96 μg mL-1 for carbidopa, respectively. The method’s accuracy was tested by adding known volume of standard solution (75 percent, 100 percent, and 125 percent concentration of the sample) to the 100 percent concentration pre-analyzed sample solution. The percentage mean recovery by standard addition experiments of levodopa and carbidopa is 99.75% and 99.55%, respectively. This method has also been successfully used for the determination of levodopa and carbidopa in pharmaceutical formulations.
Farmasötik formülasyonlarda levodopa ve karbidopanın eşzamanlı olarak ayrılması, tanımlanması ve belirlenmesi için uygun ve basit yüksek performanslı sıvı kromatografi yöntemi geliştirilmiştir. Analiz, Ace C18 (5 μm, 4.6 x 250 mm) kolonu kullanılarak gerçekleştirildi ve ayırma, UV ile 1.2 mL min-1 akış hızında pompalanan 50 mM KH2PO4 (pH 2.3) içeren bir mobil faz kullanılarak ve 280 nm’de dedekte edilerek gerçekleştirildi. Yöntem, Uluslararası Uyum Konferansı (ICH) kabul kriterleri temelinde valide edilmiştir. Alıkonma zamanlarında bu analitlere karşılık gelen hiçbir girişim bulunmadığından metod seçicidir. Her iki ilaç için alıkonma süreleri levodopa ve karbidopa için sırasıyla 2.939 dakika ve 5.018 idi. Analiz metodu valide edilmiş ve levodopa ve karbidopa için sırasıyla 25-125 μg mL-1 ve 6.25-31.25 μg mL-1 konsantrasyon aralığında doğrusal olduğu gösterilmiştir. Analiz metodu, analiz edilen konsantrasyon aralığında iyi doğrusallık (R2> 0.999) sergiledi ve gün içi ve günler arası iyi kesinlik gösterdi. Teşhis limiti (LOD) ve tayin limiti (LOQ), kalibrasyon eğrisinin eğiminin ve kaymasının standart sapmasına göre belirlenmiştir. LOD ve LOQ değerleri levodopa için sırasıyla 0.70 μg mL-1 ve 2.13 μg mL-1 ve karbidopa için 1.31 μg mL-1 ve 3.96 μg mL-1 idi. Yöntemin doğruluğu, önceden analiz edilen yüzde 100 konsantrasyona bilinen bir hacimde standart çözelti (yüzde 75, yüzde 100 ve yüzde 125 konsantrasyon) ilave edilerek test edildi. Standart levodopa ve karbidopa ilavesiyle ortalama geri kazanım yüzdesi sırasıyla %99.75 ve %99.55’tir. Bu metod, farmasötik formülasyonlarda levodopa ve karbidopa tayini için de başarılı bir şekilde kullanılmıştır.
Download Article in PDF (594.0 kB)